Introduction
PyPPL - A Python PiPeLine framework







Documentation | API | Change log
Features
- Process caching.
- Process reusability.
- Process error handling.
- Runner customization.
- Easy running profile switching.
- Plugin system.
Installation
pip install PyPPL
Plugin gallery
(*) shipped with PyPPL
- pyppl_report: Generating reports for PyPPL pipelines
- pyppl_flowchart: Generating flowchart for PyPPL
- pyppl_export*: Exporting outputs generated by PyPPL pipeline
- pyppl_echo*: Echoing script output to PyPPL logs"
- pyppl_rich*: Richer information in logs for PyPPL
- pyppl_strict*: More strict check of job success for PyPPL
- pyppl_lock*: Preventing running processes from running again for PyPPL
- pyppl_annotate: Adding long description/annotation for processes
- pyppl_require: Checking and installing requirements for processes
- pyppl_jobtime: Job running time statistics for PyPPL
- pyppl_notify: Email notifications for PyPPL
- pyppl_runcmd: Allowing to run local command before and after each process for PyPPL
- pyppl_runners: Common runners for PyPPL
Writing pipelines with predefined processes
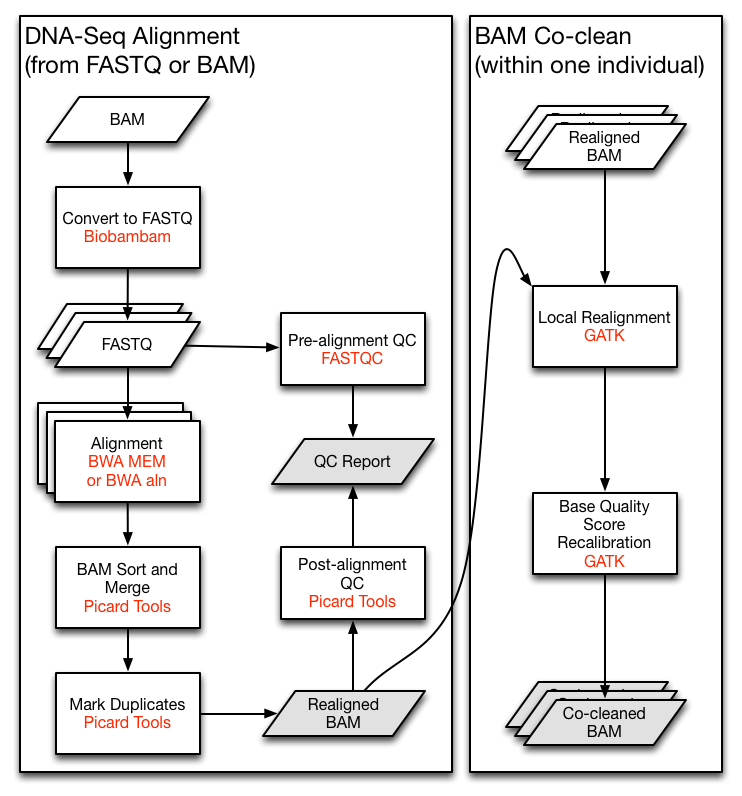
Let's say we are implementing the TCGA DNA-Seq Re-alignment Workflow (The very left part of following figure). For demonstration, we will skip the QC and the co-clean parts here.
demo.py:
from pyppl import PyPPL, Channel
# import predefined processes
from TCGAprocs import pBamToFastq, pAlignment, pBamSort, pBamMerge, pMarkDups
# Load the bam files
pBamToFastq.input = Channel.fromPattern('/path/to/*.bam')
# Align the reads to reference genome
pAlignment.depends = pBamToFastq
# Sort bam files
pBamSort.depends = pAlignment
# Merge bam files
pBamMerge.depends = pBamSort
# Mark duplicates
pMarkDups.depends = pBamMerge
# Export the results
pMarkDups.config.export_dir = '/path/to/realigned_Bams'
# Specify the start process and run the pipeline
PyPPL().start(pBamToFastq).run()

Implementing individual processes
TCGAprocs.py:
from pyppl import Proc
pBamToFastq = Proc(desc = 'Convert bam files to fastq files.')
pBamToFastq.input = 'infile:file'
pBamToFastq.output = [
'fq1:file:{{i.infile | stem}}_1.fq.gz',
'fq2:file:{{i.infile | stem}}_2.fq.gz']
pBamToFastq.script = '''
bamtofastq collate=1 exclude=QCFAIL,SECONDARY,SUPPLEMENTARY \
filename= {{i.infile}} gz=1 inputformat=bam level=5 \
outputdir= {{job.outdir}} outputperreadgroup=1 tryoq=1 \
outputperreadgroupsuffixF=_1.fq.gz \
outputperreadgroupsuffixF2=_2.fq.gz \
outputperreadgroupsuffixO=_o1.fq.gz \
outputperreadgroupsuffixO2=_o2.fq.gz \
outputperreadgroupsuffixS=_s.fq.gz
'''
pAlignment = Proc(desc = 'Align reads to reference genome.')
pAlignment.input = 'fq1:file, fq2:file'
# name_1.fq.gz => name.bam
pAlignment.output = 'bam:file:{{i.fq1 | stem | stem | [:-2]}}.bam'
pAlignment.script = '''
bwa mem -t 8 -T 0 -R <read_group> <reference> {{i.fq1}} {{i.fq2}} | \
samtools view -Shb -o {{o.bam}} -
'''
pBamSort = Proc(desc = 'Sort bam files.')
pBamSort.input = 'inbam:file'
pBamSort.output = 'outbam:file:{{i.inbam | basename}}'
pBamSort.script = '''
java -jar picard.jar SortSam CREATE_INDEX=true INPUT={{i.inbam}} \
OUTPUT={{o.outbam}} SORT_ORDER=coordinate VALIDATION_STRINGENCY=STRICT
'''
pBamMerge = Proc(desc = 'Merge bam files.')
pBamMerge.input = 'inbam:file'
pBamMerge.output = 'outbam:file:{{i.inbam | basename}}'
pBamMerge.script = '''
java -jar picard.jar MergeSamFiles ASSUME_SORTED=false CREATE_INDEX=true \
INPUT={{i.inbam}} MERGE_SEQUENCE_DICTIONARIES=false OUTPUT={{o.outbam}} \
SORT_ORDER=coordinate USE_THREADING=true VALIDATION_STRINGENCY=STRICT
'''
pMarkDups = Proc(desc = 'Mark duplicates.')
pMarkDups.input = 'inbam:file'
pMarkDups.output = 'outbam:file:{{i.inbam | basename}}'
pMarkDups.script = '''
java -jar picard.jar MarkDuplicates CREATE_INDEX=true INPUT={{i.inbam}} \
OUTPUT={{o.outbam}} VALIDATION_STRINGENCY=STRICT
'''
Each process is indenpendent so that you may also reuse the processes in other pipelines.
Pipeline flowchart
# When try to run your pipline, instead of:
# PyPPL().start(pBamToFastq).run()
# do:
PyPPL().start(pBamToFastq).flowchart().run()
.pyppl.svg will be generated under current directory.
Note that this function requires Graphviz and graphviz for python.
See plugin details.

Pipeline report
See plugin details
pPyClone.report = """
## {{title}}
PyClone[1] is a tool using Probabilistic model for inferring clonal population structure from deep NGS sequencing.
}})
```table
caption: Clusters
file: "{{path.join(job.o.outdir, "tables/cluster.tsv")}}"
rows: 10
```
[1]: Roth, Andrew, et al. "PyClone: statistical inference of clonal population structure in cancer." Nature methods 11.4 (2014): 396.
"""
# or use a template file
pPyClone.report = "file:/path/to/template.md"
PyPPL().start(pPyClone).run().report('/path/to/report', title = 'Clonality analysis using PyClone')
